
MUSICODE's M42 meeting was live on June
2 yrs ago
We are happy to welcome you to the "Summer School on Multiscale Modelling and Open Innovation Platforms", organized by the Institute of Materials Science and Computing of the University Research Center of Ioannina and hosted by MUSICODE project.
The design, processing and manufacturing of advanced devices involves a plethora of physical phenomena in materials that appear in many different length scales. These range from chemical interactions, microstructure thermodynamics, rheology, and phase changes to opto-electronic interactions in full performing devices. The theoretical understanding of all these phenomena withing a comprehensive multi-scale view is paramount for optimizing materials properties, their processing technologies, and their device functionalities and performance metrics. In this summer school, academic and industrial experts will introduce students to multiscale modelling. By the end of the course, students will have a good grasp of the fundamental physics and phenomena in each scale as well as a first understanding of the numerical methods involved in the simulations. Many examples and case studies, both in basic and applied (industrial) research, will also be presented. Lectures include:
Also included are lectures on newly established Open Innovation Platforms. These include simulation frameworks capable of handing and executing a series of multiscale simulations, where the output of one scale becomes the input for the next. Several European projects working on such Open Simulation Platforms will be presented, by the Researchers coordinating them, to give students a glimpse of all the exciting developments in Europe on materials modelling. Lectures include:
The summer school is held from 18 to 23 of July 2022 in the form of a webinar (zoom), with exam/test and certificate of participation and successful examination.
You can find the poster here: Poster
We look forward to seeing you online!

You can find the program in PDF format here: Program
 Dr. George Volonakis, The Rennes Institute of chemical sciences (ISCR)
Dr. George Volonakis, The Rennes Institute of chemical sciences (ISCR)
Dr. G. Volonakis main research activities involve the computational study of the electronic structure of solids, the materials design from first-principles of novel perovskites, and the study of the interfaces between two-dimensional materials with perovskite solar cells for achieving optimized charge extraction in solar-cells. Since January 2020, he moved to France where he holds the “Chaire de Recherche” at the Institut des Sciences Chimiques de Rennes.
 Assoc. Prof. Dimitrios Papageorgiou, University of Ioannina (UoI)
Assoc. Prof. Dimitrios Papageorgiou, University of Ioannina (UoI)
Associate Professor Dimitrios Papageorgiou of Computational Materials Science. He is specialist in (i) ab-initio electronic structure calculations, (ii) molecular dynamics and Monte Carlo techniques for organic electronicnanomaterials, (iii) non-linear optimization techniques.
 Prof. Vangelis Harmandaris, Cyprus Institute (CyI)
Prof. Vangelis Harmandaris, Cyprus Institute (CyI)
Prof. Vangelis Harmadaris research interests concern on the development of mathematical and computational multi-scale methodologies for complex molecular systems and materials of scientific and technological interest.
 Prof. Britta Nestler, Karlsruhe Institute of Technology (KIT)
Prof. Britta Nestler, Karlsruhe Institute of Technology (KIT)
Prof. Britta Nestler is interested on research on microstructure simulation in materials science. She is involved in a variety of European and national research projects, where experts from experiments and computations work together to improve and develop new materials.
 Dr. Michael Selzer, Karlsruhe Institute of Technology (KIT)
Dr. Michael Selzer, Karlsruhe Institute of Technology (KIT)
Michael Selzer (born 1976) is working in the group of Prof. Nestler and leads the sustainable software development around Pace3D. As a computer scientist, he was able to gain extensive insights into material sciences during this time, so that he was able to earn his doctorate at the Faculty of Mechanical Engineering at KIT in 2014.
 Dr. Aron Kneer, TinniT
Dr. Aron Kneer, TinniT
Dr. Aron Kneer is the Senior Project Manager of TinniT Technologies GmbH.
 Dr. Sandra Jenatsch, Fluxim
Dr. Sandra Jenatsch, Fluxim
Dr. Sandra Jenasch is the R&D Manager in Fluxim regarding Organic Electronics. Currently, she contributes to a variety of research projects of Fluxim and is also coaching Ph.D and M.Sc. students in collaboration with several European universities. Some of her responsibilities include the training of business partners and customers using the scientific tools of Fluxim.
 Dr. Urs Aeberhard, Fluxim
Dr. Urs Aeberhard, Fluxim
Dr. Urs Aeberhard is senior R&D Manager in Fluxim. Since October 2018, he joined the Winterthur based Company Fluxim as an R&D Scientist to work on the simulation of organic LED and solar cells. His research interests are: Charge carrier generation, recombination and transport in quantum wells, wires and dots, Quantum transport formalisms (especially NEGF) and applications in opto-electronics, Many-body quantum theory of photoexcited semiconductor nanostructures, Advanced concepts in photovoltaics: hot-carrier cells, multi-junction solar cell architectures, multi-exciton generation, up-/down-conversion, etc and multiscale modelling of nanostructure-based solar cell devices.
 Prof. Dr. Ing. Bořek Patzák, Czech Technical University in Prague (CVUT)
Prof. Dr. Ing. Bořek Patzák, Czech Technical University in Prague (CVUT)
Prof. Dr. Ing. Bořek Patzák main research activities are: multi-physics modelling in mechanics, particularly adaptive techniques, constitutive modelling of quasibrittle materials, high-performance computing and software development. Original author of an open source finite element simulation code OOFEM (www.oofem.org) and multi-physics integration platform MuPIF (www.mupif.org).
 Prof. Elefterios Lidorikis, University of Ioannina (UoI)
Prof. Elefterios Lidorikis, University of Ioannina (UoI)
Prof. Elefterios Lidorikis is a Professor in Materials Science Engineer department of University of Ioannina. His research interests are: computational electrodynamics and multi-scale modelling of materials, nanophotonics, plasmonics, photonic crystals, plasmonics for solar harvesting applications, optical proprties of graphene, carbon nanotubes and photonic applications and devices.
 Dr. Peter Klein, Fraunhofer ITWM
Dr. Peter Klein, Fraunhofer ITWM
Peter Klein is a Senior Scientist at the Fraunhofer ITWM, Kaiserslautern. During his 22 years at the ITWM he hold various R&D roles at several departments, covering fluid dynamics, parallelization, molecular dynamics, development of production devices and medical applications. Since his PhD thesis, he is working on multiscale models for materials, with a focus on surface and interface related phenomena.
 Dr. Natalia Konchakova, Helmholtz-Zentrum Geesthacht
Dr. Natalia Konchakova, Helmholtz-Zentrum Geesthacht
Dr. Natalia Konchakova is a senior scientist at Helmholtz-Zentrum Geesthacht, Germany. She works in the fields of materials modelling, simulation and characterization of lightweight metals, computation analysis of mechanically induced corrosion, and investigation of Magnesium based materials for bio- and structural industrial applications.
Within this series of lectures, I will go through the fundamentals of employing calculations from first-principles to calculate the electronic structure of materials. There will be a short introduction to the density functional theory and its formalism, focusing on its application to solids.  I will present its current limitations, as well as an overview of methods to improve the description of materials properties. The lecture will include some practical examples of successful applications of atomistic materials modelling to prototype materials, before focusing on the emerging class of solar-cell material: halide perovskites. I will start with a short historic overview of the materials, before discussing the electronic structure of the prototype lead halide perovskites. We will also go through the recent efforts on the front of computational materials design of lead-free perovskites that retain the same remarkable electronic and optical properties. In the last part, we will go through some rational design rules, the connection between halide perovskites and oxides, and we will briefly discuss the importance of their surface and interfaces for potential application to energy generation devices like solar-cells and photocatalysts.
I will present its current limitations, as well as an overview of methods to improve the description of materials properties. The lecture will include some practical examples of successful applications of atomistic materials modelling to prototype materials, before focusing on the emerging class of solar-cell material: halide perovskites. I will start with a short historic overview of the materials, before discussing the electronic structure of the prototype lead halide perovskites. We will also go through the recent efforts on the front of computational materials design of lead-free perovskites that retain the same remarkable electronic and optical properties. In the last part, we will go through some rational design rules, the connection between halide perovskites and oxides, and we will briefly discuss the importance of their surface and interfaces for potential application to energy generation devices like solar-cells and photocatalysts.
===============
To simulate charge transport in disordered organic electronic materials (such as semiconducting organic molecules and polymers)  we need modelling that spans across different physics and different length scales. Specifically, it requires the combination of realistic models for the atomistic structure (arrangements, domains), the electronic interactions (site energies, transport parameters) and stochastic processes (charge hopping). A proper modelling scheme follows a hierarchical multiscale approach including quantum mechanical (by Density Functional Theory), classical atomistic (by Molecular Dynamics) and stochastic electronic transport (by Kinetic Monte Carlo) simulations. After an intuitive and educational introduction to the modelling methods, we’ll look at the details and examples of charge transport in organic semiconductors typically utilized in organic photovoltaics.
we need modelling that spans across different physics and different length scales. Specifically, it requires the combination of realistic models for the atomistic structure (arrangements, domains), the electronic interactions (site energies, transport parameters) and stochastic processes (charge hopping). A proper modelling scheme follows a hierarchical multiscale approach including quantum mechanical (by Density Functional Theory), classical atomistic (by Molecular Dynamics) and stochastic electronic transport (by Kinetic Monte Carlo) simulations. After an intuitive and educational introduction to the modelling methods, we’ll look at the details and examples of charge transport in organic semiconductors typically utilized in organic photovoltaics.
===============
Macromolecular systems (polymers) are characterized by a very broad range of characteristic time and length scales; for example, even a single polymer  chain exhibits length scales ranging from the bond length (~ Å) to the size of the chain (O(10 nm)) and corresponding time scales ranging from a few fs (10-15¬ sec) for the bond vibrations up to the order of milliseconds or even seconds for the whole chain relaxation. For this reason, the computational study, and the prediction of structure-property relations, of polymer-based materials is a very challenging, and intense, research field. Due to the above spectrum of spatiotemporal scales, a systematic combination of simulation approaches, describing a specific system in different levels, is necessary.
Within this series of lectures, we are going to discuss a hierarchical computational methodology across different scales, involving all-atom and mesoscopic (coarse-grained, CG) dynamic simulations for polymer-based materials. Atomistic molecular dynamics simulations offer a direct approach for the detailed study of systems, in the classical approximation. However, due to the computational costs it is not feasible to take into account all the atomistic degrees of freedom in long, high molecular weight, polymer chains. To reduce the dimensionality of the model systems, and increase the length and time scales accessible by simulations, coarse-grained models that describe group of atoms as “superatoms”, or “particles”, can be used.
chain exhibits length scales ranging from the bond length (~ Å) to the size of the chain (O(10 nm)) and corresponding time scales ranging from a few fs (10-15¬ sec) for the bond vibrations up to the order of milliseconds or even seconds for the whole chain relaxation. For this reason, the computational study, and the prediction of structure-property relations, of polymer-based materials is a very challenging, and intense, research field. Due to the above spectrum of spatiotemporal scales, a systematic combination of simulation approaches, describing a specific system in different levels, is necessary.
Within this series of lectures, we are going to discuss a hierarchical computational methodology across different scales, involving all-atom and mesoscopic (coarse-grained, CG) dynamic simulations for polymer-based materials. Atomistic molecular dynamics simulations offer a direct approach for the detailed study of systems, in the classical approximation. However, due to the computational costs it is not feasible to take into account all the atomistic degrees of freedom in long, high molecular weight, polymer chains. To reduce the dimensionality of the model systems, and increase the length and time scales accessible by simulations, coarse-grained models that describe group of atoms as “superatoms”, or “particles”, can be used.
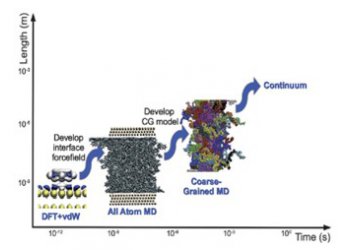
We describe systematic “bottom-up” CG models, which are obtained by applying data analytics approaches on large datasets (configurations) obtained from atomistic simulations, providing also error estimates for the derived CG models. In addition, we present machine learning based approaches for linking different scales, by re-inserting atomic detail in CG configurations.
The systematic combination of the above approaches allows the study of macromolecular systems, of high molecular weight, over a broad range of time scales, from a few fs up to ms. and the prediction of their (structural, dynamical, rheological, etc.) properties. As examples, we present results concerning the properties of various polymer-based materials; from polymer melts, up to polymer thin films and graphene-based polymer nanocomposites.
===============
Phase-field modelling has become a fairly versatile technique for the treatment of microstructure formation and phase transition problems. It usually operates on a mesoscopic length-scale exploring microstructural characteristics at a micrometer resolution. With this scope, the method serves as the bonding chain between atomistic and macroscopic simulation schemes. Herewith, the phase-field method plays a central role in multi-scale materials modelling and naturally desires the exploitation of large representative volume elements. Furthermore, the combination of phase-field modelling with multiphysics applications such as heat and mass transfer, continuum mechanics, fluid flow, micromagnetism and electrochemistry has been achieved. Incorporating multiscale and multiphysics, phase-field modelling is central for the future technology: Integrated Computational Materials Engineering (ICME)’’ as it allows information transfer between both, experimentalists and modellers as well as between different materials modelling methods. 
In this lecture, we present the fundamental formulation of the phase-field model, show an extension to multicomponent and multiphase material systems and discuss techniques to efficiently transfer thermodynamic databases to provide direct access to the Gibbs free energies of the different phases. In order to prepare Gibbs energy functions as input information for the phase-field simulations, a semi-automated programming interface to generate approximated or precise mathematical expressions of the Gibbs energies based on Calphad data is introduced for binary and ternary material systems. The formulations are verified by recalculating the equilibrium concentrations of the phases and rebuilding the phase diagrams in the considered concentration and temperature ranges, prior to the simulation studies. For all comparisons, a close match is achieved between the results and the Calphad databases.
Alloy systems with three or more chemical species can form a broad variety of different microstructures depending on physical parameters and processing conditions. To investigate the diversity of pattern formations, a full three dimensional modelling is mandatory and requires intense computational power. Computations of large scale 3D microstructures coupled with multiphysical effects essentially require to comprise modern multigrid and parallel algorithms to efficiently use high performance clusters. A multiphysics microstructure simulation framework Pace3D is being developed since more than 20 years and offers a parallelized simulation environment for large applied calculations on high-performance computers with excellent scalability properties and extensive data analysis algorithms for the virtual and accelerated design of new materials and for the optimized design of process flows. We choose a ternary eutectic alloy to demonstrate the power of high performance computations for describing the physical mechanisms of experimentally observed phase ordering during solidification and to derive morphology transition diagrams. Using advanced data analysis tools and principal component algorithms, we illustrate the necessity of massively parallel high performance computing techniques to resolve the microstructures in sufficiently large representative volume elements. Furthermore, we choose selected highlights of computed 3D microstructures to illustrate the broad applicability to study e.g. wetting phenomena of immisible and compound droplets on flat, porous and chemically structured substrates. By combining the phase-field method with elasto-plastic models, we show a detailed view into the stress-strain evolution and crack propagation in polycrystalline grain structures. Simulations allow for the first time the in-situ insight not only into the final microstructure, but also in the three-dimensional structure formation process.
===============
During material processing porous media is resulting if phase separation of poly- mers happens and evaporation of solvents occur. The material coming out from such a process is cellular which means that the solid phase does not fill complet- ley a volume. Porous media which is flown through or even if the pores are filled by another material which can be gas, liquid or another phase which is solidifi- cated after the filling process. Welknown examples for such kind of porosities are membranes, foams, textiles and also a bet of grains. Flow through a porous region is driven by capillary forces or any pressure load. The flow pattern is complex due to different pore sizes and obstacles represented by the solid phase therefore computational fluid mechanics tools are needed to predict the flow behavior. Furthermore heat may be conducted through this porous system and a part of this energy is stored inside the solid phase. To be able to compute huge domains filled with a porous material and another media (e.g. heat storage system - metal foam and paraffin) the micro structure can not be resolved for a computation because of the immense computing power needed. There exist another approach to address a porosity within a computational model. This model bases on a porous region description which allows to define the flow re- sistance as a tensor for this region. This tensor describes the friction losses of a porosity and can be defined also for anisotropic porosities. The other thermal properties can be defined in the same way. The only point is that these data represent effective values for the computational domains and do not reflect local velocity dependend effects. In the case of heat transfer this averaging procedure leads to a wrong prediction of the heat transfer process in a porous region. An interesting extension to a CFD solver is the dual-porosity approach which is based on an additional energy equation for the solid which is not represented (ressolved) in a porous region. The dual-porosity approach bases on the de- scription of a mobile (fluid) and a immobile (solid) phase of a porous region whereas for the immobile phase (e.g. matrix of a membrane) this additional energy equation is solved. The two energy equations (fluid & solid) are coupled by locally computed heat ransfer coefficient which depends on the local velocity and temperature distribution. The porosity defines the portion of the fluid phase in each computational cell. Within this course the dual-porosity approach will be introduced and the theorie based on the two-equation model will be explained. Furthermore, the advantage of this method compared to the classic porosity approach is shown on the basis of various practical examples.
Computational fluid dynamics (CFD) is an established method to compute in a two or three-dimensional domain the flow field. The numerical solution of the Navier-Stokes equations is based on the discretization of the conservation equations. A common discretization scheme is the finite volume method. The simulation domain is therefore discretized by finite volumes. The discretized conservation equations are partial differential equations which are nonlinear. Fluid mechanics is a subfield of continuum mechanics. If the absolute pressure is reduced, the viscous flow changes to molecular dynamic movement processes. In order to be able to calculate near-vacuum flows despite the restriction im- posed by the continuum-mechanical considerations, the CFD method has to be extended to be applicable for this phyical condition. An example of these ex- tensions is the modification of the non-slip condition on the walls. Due to the low density, the fluid experiences a changed viscous friction inside the fluid as well as on the walls, so that a slip condition to the walls has to be defined as a function of the free path length of the molecules. Furthermore, the material properties such as viscosity, thermal conductivity, diffusion coefficient, etc. must be calculated by functions depending on the Lennard-Jones length and energy, so that the numerical fluid mechanics method can be applied to this kind of physics. This course deals with the basics of fluid mechanics and in particular with the necessary extensions of the CFD technology in order to demonstrate flow processes under low-pressure conditions using the example of the deposition of organic molecules on a substrate.
===============
This first part starts with the introduction of the theoretical formalism that is used to describe the physical processes central to the operation of opto-electronic devices based on thin organic or metal halide perovskite films. Special consideration is given to the role of disorder and excitonic behavior for charge generation/recombination, transport, and extraction/injection in organic opto-electronics, to the mobile ionic species in perovskites, and to the coherent thin film optics required to capture cavity effects in absorption and emission properties. Also, the assumptions made in the transition from an atomistic to a continuum formulation of the material are discussed. This leads to the formulation of a comprehensive modelling framework based on the coupling of continuity equations with drift-diffusion currents for free charge carriers and mobile ions to Poisson’s equation, the exciton diffusion equation (for organics), and the optical models for absorption and emission. The specific application of the framework is elaborated on for prototypical cases of steady-state, transient and frequency domain simulations of single junction and tandem devices. Finally, an outlook is given on how to use the insight from device level modelling for the simulation-based assessment of opto-electronic performance at larger scale, i.e., of PV modules and OLED panels.
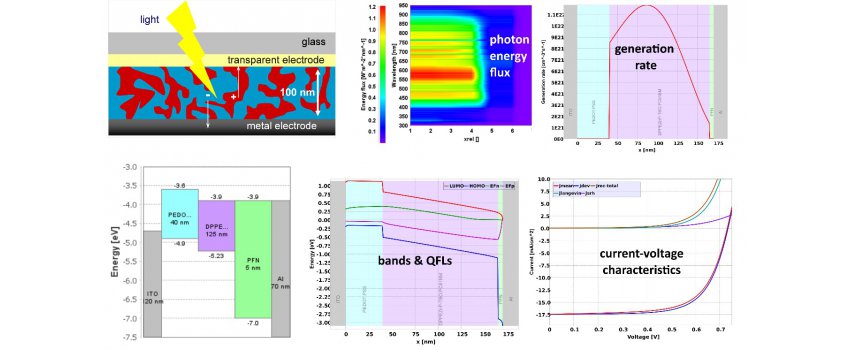
In the second part of the lecture focusses on different application examples. We will start with an excursion to opto-electrical device measurement techniques for OLEDs and solar cells and present how those can be used to qualitatively understand device limitations. While a combination of steady-state, transient and impedance techniques are useful to pinpoint differences between sample A and B, their full potential is only exploited when combined with device simulations. We will present how this combination can be used to determine reliably material and device parameters, understand microscopic degradation mechanisms, and even optimize active layer compositions. Lastly, examples of large area simulations are presented that allow us to quantify upscaling losses and detect defects in large area solar cells and modules.
===============
Tailoring and optimizing the properties of complex materials for specific applications leads to increased performance and significant cost savings. The ability to create a digital “virtual twin” by means of modeling of a real problem makes the selection and optimization of new products faster and cheaper than traditional approaches based on experience and prototype evaluation. This opens up new opportunities for exploring the untapped potential of new materials.
Consequently, modeling is becoming essential to the design and optimization of a complex material. Complex materials exhibit different features at varying time and length scales with properties highly influenced by different manufacturing processes. There is a need for reliable multi-scale and multi-physics models and workflows allowing the simulation of composites materials, including the manufacturing process. Today, many advanced single purpose models are available that fit into the concept. Therefore, from the practical and economic point of view, the focus is on the re-use of the existing tools and their integration. However, the existing models use different modeling approaches and rely on specific data formats, which present another level of challenge and complexity, from the software engineering perspective. 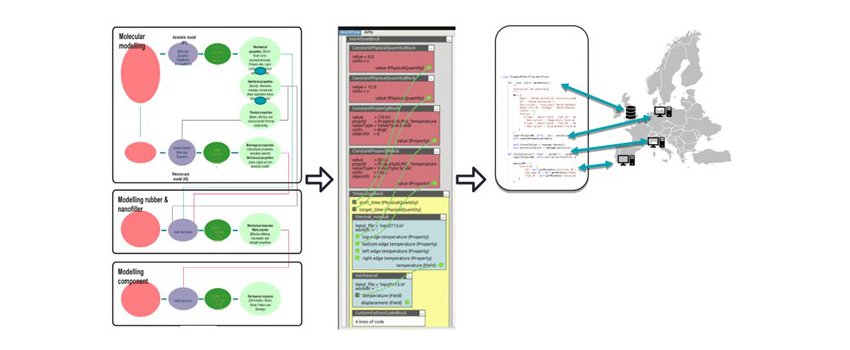
Thus, there is a strong need for integration platforms enabling documented, automated, and repeatable inter-operable integration of existing models and data sources into simulation workflows. Interoperability, by definition, implies standards. Traditional approaches build on syntactic interoperability, implying specific communication protocols and data conversions. Today, the most attractive approaches are based on semantic interoperability, where data and models are brought together with their meanings, which then allows for automated machine interpretation, translation, data manipulation, and verification.
In the lecture, the generic approaches to interoperability will be presented and discussed. In the second part, the design of a distributed, open-source simulation platform MuPIF, developped in the frame of past and present EU funded projects will be presented and demonstrated on real scale examples. Finally, the benefits of employing traceable, interoperable modeling environment for material research will be discussed.
===============
MUSICODE addresses the H2020 Call DT-NMBP-11-2020 “Open Innovation Platform for Materials Modelling” with the aim to create a comprehensive modelling environment for materials design, materials processing, and device optimization in the Organic Electronics application domain. The platform integrates: (a) modelling workflows spanning the micro-, meso- and macro-scales, (b) graphical user interface tools for workflow design, (c) data management and execution framework with ontology-based semantic interoperability, (d) plug-ins to Materials Modelling Marketplaces, Open Translation Environment, HPC infrastructures. Industry workflows for optimizing material properties and manufacturing will be demonstrated.
===============
 The H2020 funded project VIPCOAT (Grant Agreement 952903) is focused on the establishment of an Open Innovation Platform for active protective coatings. Multi scale materials modeling and business decision support tools assist industry to invent and manufacture optimal coating system for self-protecting paints in aviation industry. Moreover, the platform will support business to monitor technological requests/ KPIs and environmentally friendly requirements/ limitations during development project execution.
The H2020 funded project VIPCOAT (Grant Agreement 952903) is focused on the establishment of an Open Innovation Platform for active protective coatings. Multi scale materials modeling and business decision support tools assist industry to invent and manufacture optimal coating system for self-protecting paints in aviation industry. Moreover, the platform will support business to monitor technological requests/ KPIs and environmentally friendly requirements/ limitations during development project execution.
A general overview and the main objectives of VIPCOAT will be presented in the lecture. Additionally, initial steps towards coating industry digitalization and data/knowledge generation for the topology of active protective coating will be demonstrated. Moreover, an example of modeling and simulation of corrosion inhibitors leaching from a coating matrix to defect area will be considered [1]. The delivery of corrosion inhibitor molecules from active pigments can occur via diffusion through the polymer matrices after taking up water or via a percolation network formed over time as a consequence of pigment dissolution.
![Microstructure of a protective coating [2]](/images/c/2/9/c/9/c29c9ca33514b6e3d5fc7cb6c34bbdc77aeedf59-vipcoat.png "Microstructure of a protective coating [2]")
[1] J. Höller, J. Niedermeyer, C. Redenbach, N. Ecke, A.K. Schlarb, H. Andrä, P. Klein, Heat Mass Transfer 56, 2020.
[2] J.S.Laird, P.Visser, S. Ranade, A.E. Hughes, H. Terryn, J.M.C. Mol, Progress in Organic Coatings, 134, 2019.
===============
===============
To register for the summer school, please download and complete the registration form and return to the school email musicode@uoi.gr along with a copy of the bank receipt. Your registration will not be considered final until we receive the bank receipt.
Registration fees
The registration fee of 50 euros provides access to all courses and course material. A few fee waivers will be offered to students based on merit. To be considered for a fee waiver please send us your current CV and a letter of reference. The registration deadline is extended. The new deadline is at 30th of June.
Method of payment is bank transfer. Please use the following account:
Please include "NAME_83167", where NAME is the participant last name, as the reason for the transaction.
Any bank transfer expenses should be covered by the participant.

![]()
This project has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No 953187

